Introduction
The biopharmaceutical industry has been growing and evolving at a pace that’s hard to match, especially in terms of manufacturing. Gone are the days when developers looked at monoclonal antibodies (mAbs), or antibodies that are made by identical immune cells cloned from a single cell, as a challenge – rather, mAbs have become an industry standard, with well-established research, development, and manufacturing processes that have proven to be robust and scalable. In the current biopharmaceutical landscape, developers are constantly encountering new and diverse biological molecules with complex structures. While these novel biologics are filling drug pipelines, they are also challenging existing manufacturing processes, both in upstream, downstream and analytical development.
Understanding what challenges these diverse molecules can create and how to manage these challenges is key to ensuring they reach patients safely and efficiently.
A growing molecular diversity
Biologics have now claimed a significant chunk of the drug development pipelines in most pharmaceutical companies. Although they are not a new class of drugs, recent technological advancements and our expanding knowledge of human biology have driven growth and interest in this field. Experts think new biologics have great potential to treat unmet medical needs, due to their complicated structures and the resulting specificity in target-binding and efficacy.1, 2
Biopharmaceuticals are typically defined as drugs produced from living organisms or drugs that contain components of living organisms. Aside from mAbs, there are many other classes of biologic drugs,3 including alternative antibody types (antibody fragments, antibody-drug conjugates, bispecific antibodies), recombinant proteins (e.g., hormones such as insulin and human growth hormone), cell therapies (e.g., CAR T cells, stem cell therapies), and viral vectors (certain vaccines and gene therapies).
Next-generation antibody derived molecules and viral vectors have overcome some of the therapeutic limitations of simple small-molecule drugs and mAbs, but on the flip side, they have created a few hurdles in process development.
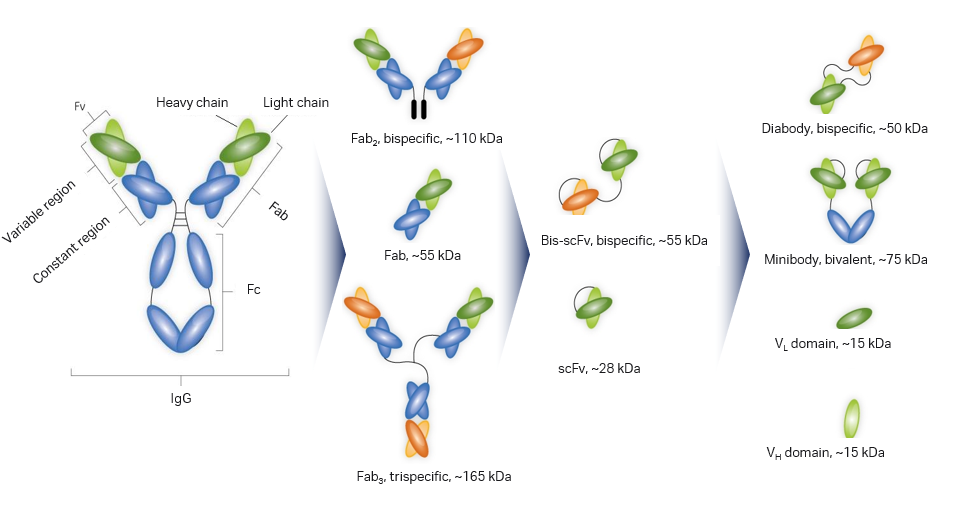
Fig 1. A schematic showing an example of the variety of molecular diversity when it comes to antibody derived biopharmaceuticals.
The challenge of developing a scalable industrial process
Many of these molecules originate in academic settings, where innovation has led to groundbreaking discoveries for drug development. However, academic labs are not typically set up for optimizing process development or improving manufacturing efficiency. Additionally, basic research is not always carried out with regulatory acceptance in mind.
As such, translating these ground-breaking discoveries into scalable processes has become a top challenge for biomanufacturers pursuing novel therapies. “In my opinion, one of the biggest challenges we are faced with as developers, is scaling up production of new molecules and ensuring the process delivers a high enough yield of quality drug substance for clinical trials and commercial programs,” says Dr. Mats Lundgren, customer applications director at Cytiva, who has more than two decades of experience in vaccine and viral vector processing.
Also, some of these early stage production processes are based on legacy technologies. This can lead to processes which are not easily scalable or economical. For instance, the use of bovine serum as a cell culture supplement for viral vectors may be commonplace in academic labs, but this practice would not work out in biomanufacturing where strict regulations on contamination and patient safety prevent animal-derived components from being used. Another example is the use of centrifugation or size-exclusion chromatography for purification purposes. While these techniques work well for small-scale processing, they are not suitable for large-scale manufacturing. The takeaway from all this, Lundgren says, is to implement technologies at the very beginning that are amenable to scale-up – but more on this later.
Particularly for the production of viral vectors, one other bottleneck is the availability of plasmid DNA. Viral vectors are viruses developed through DNA recombination techniques for delivering genetic material into target cells. They are often produced through transfection of cells with plasmid DNA (a small, circular, double-stranded DNA molecule that is distinct from a cell's chromosomal DNA and can replicate independently) containing the gene of interest. The more viral vector-based therapeutics are being produced, the greater the need for a steady plasmid DNA supply, as they are critical raw materials. But the problem here is, plasmid DNA must be manufactured and purified in a good manufacturing practice (GMP) facility or at least a high-quality plasmid DNA production area. So, scaling up this kind of operation has put a lot of pressure on drug developers to improve their production capacity to meet the rising demand for high-quality plasmid DNA.4
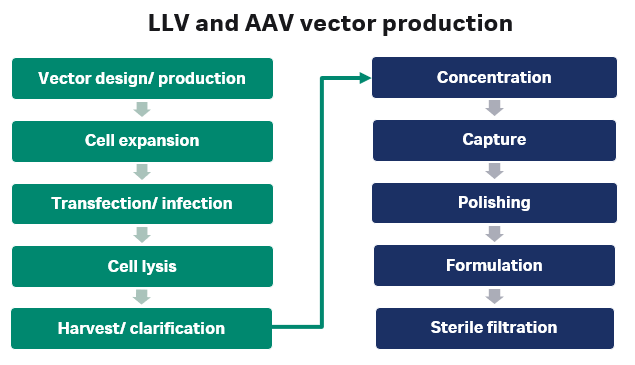
Fig 2. A schematic showing how lentiviral (LV) and Adeno associated virus (AAV) viral vectors are made.
Shorter time windows, lack of platforms, and purification challenges
With timelines for development becoming shorter and shorter, efficiency in process development has become a necessity in order to be successful. “If you look back at when the mAb field started, you had several years to work on process development,” notes Lundgren. “But nowadays, you have to do it in a very short time window, despite the fact that these are much more difficult molecules.”
Additionally, new molecules don’t come with a roadmap to development; there are no platform approaches in place yet, as with older mAb programs. Lundgren points to the novelty of viral vector-based vaccines, an entirely new field that has opened up in the last few years. Researchers and developers need to spend a significant amount of time and effort to find the best approaches for production and how to apply them. The huge variety of molecules, even within a niche field like viral vectors (adeno viruses, adeno-associated viruses, lentiviruses, retroviruses, etc.) further complicates the matter.
On the other hand, drug quality requirements are also increasing, as some new molecular formats introduce new impurities that need to be reduced. Especially when a certain molecule may appear in multiple variants, there is a responsibility to show that none of those variants affect safety and efficacy of the therapy, or if they do, there are quality control measures in place to manage/reduce them.
For example, take bispecific antibodies – multiple variants of the antibody might be formed during the expression. It’s possible that developers end up with homo- instead of heterodimerization of heavy and light chains, or random binding of light chains to heavy chains. The result would be a lower yield compared to mAb production. According to Dr. Andrew Tustian, an associate director at Regeneron in the US, one of the main pain points in purifying bispecific antibodies is the increased level of impurities. He says that the impurities are often very similar to the target molecule, making it hard to separate and remove them.5,6
Recommendations for overcoming process development challenges with new biomolecules
According to Lundgren, the first and most important step in overcoming process development challenges is to do a thorough and careful feasibility study. This would involve calculating how much material should be produced and what technologies should be used in order to achieve the necessary outcome. “You need to know your market and the technologies quite well, and you need to do your homework, so you can make a successful product at the required budget,” he says.
The next step is to use relevant process development tools in order to find the optimal process and protocols, for instance, by using the Quality by Design (QbD) approach. In this regard, Lundgren thinks a lot can be learned from how the field of monoclonal antibodies has developed over the years. A case in point is the use of cell culture components suitable from a regulatory perspective, such as using chemically defined cell culture media, instead of animal-derived serum. Chemically defined media require that all of the components must be identified and have their exact concentrations known. Implementing the use of chemically defined media is a ‘good cell culture practice’ (GCCP) and it helps develop a common standard for in vitro methods.7
The mAb field is so mature that animal-derived components are not used even in research and development. But for newer biomolecules coming from academia, that’s not the case. Academic researchers are not always familiar with regulatory constraints and they tend to stick to traditional methods used over years. Lundgren explains that there is sometimes also a misconception about getting lower yields with chemically defined cell culture media, which is not true in the present day. “We have pretty good, pure cell culture media that don’t contain animal-derived components, and they still work very well,” he states. Working with a supplier who has expertise in this area can help avoid risks associated with unsuitable raw materials while also achieving high titers.
Process development challenges with new molecules can be largely overcome by implementing scalable technologies, such as chromatography, from the get-go. One example is using a multimodal chromatography resin like Cytiva’s Capto Core 700, which can help increase speed and improve viral vectors’ process economy.8 Another example is investing in single-use technologies and equipment that reduce cleaning requirements and improve batch turnaround times.8 For scale-up of plasmid DNA manufacture, Cobra and Cytiva recently collaborated on the development of a chromatography resin ‘Capto PlasmidSelect’ that improves scalability and efficiency while also sustaining an acceptable cost of goods.4
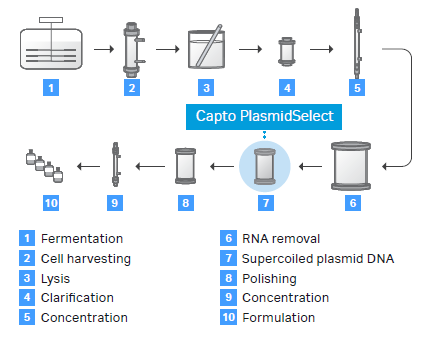
Fig 3. A schematic showing an example of a process for production of high-quality supercoiled plasmid DNA from fermentation to formulation.
Importance of analytics and characterization of the product
Investing in good analytical tools is a must during process development, because high-quality analysis is especially complicated with complex biologics, but also extremely critical. “Without good analytics, you will never be able to optimize your process because you don't really know what you're measuring,” Lundgren emphasizes. “The exact tools to be used depend on the molecule, but having a solid portfolio of different analytical tools that are suitable for your molecule is very important.”
In the case of viral vectors, many analytical methods are used to confirm different aspects such as identity, potency, purity, safety and stability. Physical viral titer (quantity of virus in a given volume of fluid) can be calculated by DNA hybridization, real-time PCR (qPCR, ddPCR), SPR technologies such as Biacore or high-performance liquid chromatography (HPLC), while functional viral titer can be measured by plaque-forming or fluorescence foci assays9, 10 Impurities can be detected by mass spectrometry or electron microscopy, and safety checked via Southern blotting, qPCR, or in vitro/in vivo assays. Microscopy, potentiometry, and osmometry are techniques used to establish stability of the vectors.9
It’s clear that expertise in a wide variety of analytical techniques is required to detect and characterize biopharmaceuticals and their impurities (both product- and process-related impurities). For most of these newer, more complex molecules, customized assays must be developed, keeping in mind speed and robustness.
Conclusion
Navigating the development challenges of biopharmaceuticals is a process that benefits from implementing scalable technologies and following QbD principles. It's vital to invest sufficient time and resources into proper, scalable process development to avoid difficulties and unnecessary costs later on. Lundgren advises developers to model their process economy early on, “It’s important to do those simulations early when you are developing your project and the business concept. We have seen examples of companies who have not done so, and unfortunately ended up in a position where it's not economically viable to produce the molecule.”
Further reading
Approaches to improve efficiency in biopharmaceutical process development
Considerations for improving outcome in biopharmaceutical process development
References
1 Jalal, B Realizing the Promise of Biologics. Harvard Health Policy Review, 2017.
2 Oo, C., Kalbag, S.S. Leveraging the attributes of biologics and small molecules, and releasing the bottlenecks: a new wave of revolution in drug development. Expert Review of Clinical Pharmacology, 2016, 9:6, 747-749.
3Biological Therapies for Cancer. National Cancer Institute, 2018.
4 Charlesworth, J., Hitchcock, T., Tharia, H. et al. Purifying plasmid DNA using a modern chromatography resin. Cytiva Bioprocessing Knowledge Center.
5 Westman, D. Bispecific antibody purification: insights and case studies. Cytiva Bioprocessing Knowledge Center, 2019.
6 Tustian, A.D., Endicott, C., Adams B. et al. Development of purification processes for fully human bispecific antibodies based upon modification of protein A binding avidity. mAbs, 2016, 8(4): 828-838.
7 van der Valk, J., Brunner, D., De Smet, K. et al. Optimization of chemically defined cell culture media – Replacing fetal bovine serum in mammalian in vitro methods. Toxicology in Vitro, 2010, 24, 1053-1063.
8Darby, N., Schmidt, S.R., Lidén, P. et al. . Preparing for the future–visions and insights for biomanufacturing. Cytiva Bioprocessing Knowledge Center.
9 King, D., Schwartz, C., Pincus, S. et al. Viral Vector Characterization: A Look at Analytical Tools. Cell Culture Dish, 2018.
10 Kramberger, P., L. Urbas, Strancar, A. Downstream processing and chromatography based analytical methods for production of vaccines, gene therapy vectors, and bacteriophages. Human Vaccines & Immunotherapeutics, 2015, 11(4): 1010-21.