For developers of cell and gene therapies (CGT), translating a drug from a biological concept to a scalable and manufacturable treatment can be the largest challenge in achieving commercial success. This is especially difficult in the CGT field, because there is a limited number of qualified personnel with both biological and process engineering know-how. Other challenges are evolving regulatory constraints and aggressive timelines from investors.
Even for experienced teams, it can be tricky to balance the efforts of reaching the first clinical trial using a manual, open method with building a more commercially suitable process. To accelerate their clinical and commercial programs, companies are choosing to work with contract development and manufacturing organizations (CDMOs). In this article, we highlight key areas where CDMOs can help companies get started in the world of CGT manufacturing. We also discuss when to engage with CDMOs to maximize commercial and clinical success.
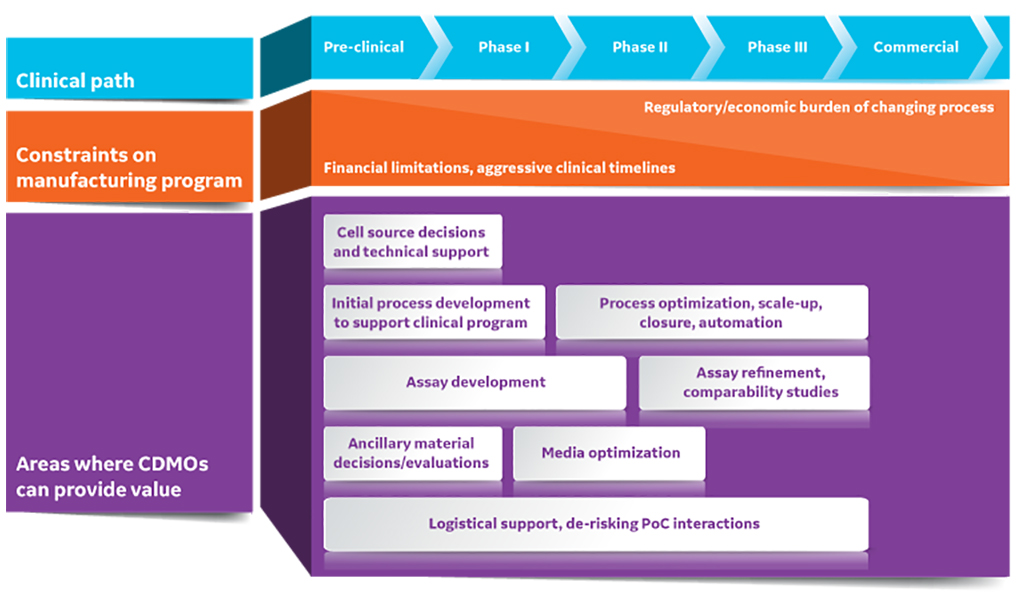
Fig 1. CDMOs can provide substantial value to early-stage cell and gene therapy developers. PoC is point of care.
Cell source considerations
CGT products start with either primary or modified cell lines as the cell source. Drug developers often choose primary cell lines when dosing requirements and patient numbers are low or when personalized, autologous therapies are produced. They typically select modified cell lines when larger patient populations are targeted or where the cells are being used as a producer line for secreted products, such as exosomes or viral vectors. Each cell source has a different set of technical, regulatory, access, and licensing considerations. A CDMO can help an early-stage CGT developer understand the limitations of their source cells. And the CDMO can aid in developing strategies around processing tissue, generating cell banks, or performing cell line engineering.
Engaging early with an experienced CDMO can also help navigate the evolving and nebulous regulatory landscape around CGT cell sources. Commercially available cell sources and vectors, though often available for unrestricted use in academic and research settings, might carry licensing fees for clinical and commercial use. In addition, primary cell and tissue sources can be very challenging to source for preclinical development. CDMOs can leverage existing relationships with public and private entities to help in sourcing these materials.
Regardless of the source, it is critical to thoroughly characterize the identity and quality of the starting cell material in preclinical development. CDMOs can tailor process and assay development programs around the unique needs of the biological input material.
Process scales and changes
In the research stage of a new CGT, cell manipulations are manual, technically complicated, and demonstrated only at laboratory scale for proof-of-concept and preclinical studies. Turning this process into one that will be robust and cost-efficient enough requires a strategic approach to scaling with diligent planning and proper documentation. Experienced experts can guide the process to avoid the pitfalls and prevent delays in time to market.
CDMOs have access to, and experience with, varied solutions that allow companies to evaluate different platforms and assess different technologies for process development (PD). Working with a CDMO at this stage can help alleviate capital costs and training burden of performing these PD studies in house.
Process changes between clinical trial phases are costly and time consuming to develop. For CGT developers, outsourcing the engineering aspects of process optimization can save time. At each development stage, equipment, methods, and materials might require process optimization or recreation. Also, changes can result in process variability. CDMOs can oversee the alignment of changes with aggressive clinical programs and assure process variability is understood.
CDMOs can also help identify the biggest value-add process modifications and recommend a step-wise program for development, optimization, and comparability studies. One point to consider is understanding where the greatest sources of risk or variability are in the current process; another is changing process steps that are not compatible with target scales. For example, a suspension-based solution might be better for commercial production where an existing laboratory process uses adherent cell types.
Analytical methods and assay development
Analytical methods are needed for raw material characterization, in-process testing, and quality control (QC) release of final products. When starting out, it can be difficult to know what the critical quality attributes (CQAs) of the drug product will be. So CGT developers often aim to capture as much data as possible about their product during process development. Identifying the correct method for a measurement is a challenge, and one option for the field is to adopt some of the best practices from design controls used in the development of medical devices and in vitro diagnostics. These include defining design inputs of a method by building a user requirements specification for each analytic, so that the assay chosen is truly fit for purpose. Examples of specifications to consider are sensitivity, specificity, turn-around time, cost per test, and the robustness and ruggedness of the assay. Working with CDMOs who have expertise in assay development and qualification can guide the choice of the appropriate specifications. Leaning on established methods at CDMOs, or transferring existing methods for further development and qualification, can also be a strategy to start a small project with the CDMO to determine whether they are the right partner.
Ideally, a company would use the same qualified analytical methods from early process development to commercial manufacture. This ensures consistency and comparability of data throughout the life cycle of the CGT product. Also, it is important to have robust assays for CQAs in place before starting major process changes, as use of these assays will serve to ensure that manufacturing changes are not having an adverse impact on the final product. Changes in methods between different phases of clinical trials – whether it is because a method is too challenging to run in a QC lab, a critical reagent is no longer available, or the method is no longer relevant based on clinical data – require lengthy and costly equivalency studies to be carried out. Having an established relationship with a CDMO, in which key assays can be run consistently and comparably to other sites, also allows for both efficient comparability studies and smoother tech transfer.
Organized and documented technology transfer
Effective, efficient technology (tech) transfer strategies assure processes are moved to a CDMO’s environment successfully. Tech transfer is a complex undertaking that requires communication and collaboration between CGT developers and the CDMO. Transparency is a key factor. Understanding a CDMO’s requirements up front helps to set clear expectations.
When working with a CDMO, tech transfer is not just a hand-off — it is the beginning of an ongoing relationship. It is important to establish a strong, cooperative rapport with the main contact at the CDMO and to maintain quality oversight on an ongoing basis to facilitate the smoothest possible transmission of knowledge. CGT developers should plan to engage in the tech transfer process throughout, to assure both parties are documenting plans and developments in detail.
The goal of tech transfer documentation is to improve the effectiveness, efficiency, quality, and compliance of the biomanufacturing tech transfer process. A thorough, reliable tech transfer methodology will go a long way towards assuring success both in technical aspects and in building the business relationship.
Tech transfer documentation serves as a critical guide, forming the basis from which standard operating procedures (SOPs) are developed. This transfer roadmap outlines important parameters of a process and product, including materials and analytical procedures and critical quality attributes (CQAs). The more defined the process specifics, the more effective this document can be in assuring a smooth transfer of technology. This documentation will also be critical should the CGT team choose to bring a process back in-house for commercial production.
Analytical methods and assay development
Working with CDMOs who have expertise in assay development and qualification can assure appropriate specifications are incorporated. Leaning on established methods at CDMOs, or transferring existing methods for further development and qualification, can also form the basis of small projects to determine if a particular CDMO is the right partner.
Analytical methods are needed for raw material characterization, in-process testing, and quality control (QC) release of final products. When starting out, it can be difficult to know what the CQAs of a drug product will be. CGT developers often aim to capture as much data as possible about their product during process development.
Identifying the correct method for a measurement is challenging. One option is to adopt best practices from design controls used in medical device development and in vitro diagnostics. These methods include defining design inputs by building a user requirement specification for each analytic, so that the assay chosen truly fits the intended purpose. Examples of specifications to consider include sensitivity, specificity, turn-around time, cost per test, and the robustness of the assay.
Ideally, a company would use the same qualified analytical methods from early process development to commercial manufacture. This ensures consistency and comparability of data throughout the life cycle of the CGT product. It is important that robust assays for CQAs are in place before initiating major process changes. This is true because use of these assays will serve to confirm that manufacturing changes are not having an adverse impact on the final product.
Changes in methods between different phases of clinical trials – whether it is because a method is too challenging to run in a QC lab, a critical reagent is no longer available, or the method is no longer relevant based on clinical data – require lengthy and costly equivalence studies. Having an established relationship with a CDMO that runs key assays consistently to other sites allows for efficient comparability studies and facilitates smooth tech transfer.
Ancillary materials
To reduce the risk of transmissible diseases and contamination in CGT products, there has been a concerted push toward the use of xeno-free (XF) reagents, animal-derived component free (ADCF) materials, and pre-sterilized single-use disposables with closed access ports. These choices can also affect cell quality, as supported by a recent example showing that serum-free media might improve potency and transduction of CAR T cells both in vitro and in vivo compared to serum-containing media (1).
Many ancillary material manufacturers are now offering reagents and consumables to support CGT developers. These materials can be up to ten times more expensive than their research use only (RUO) counterparts. And they often suffer from longer manufacturing and lead times. Therefore, CGT developers should identify and budget for good manufacturing practices (GMP)-compliant reagents and consumable sources early on. Leveraging CDMOs at an early development stage is helpful, as they can establish primary and secondary suppliers of reagents and consumables. This partnership can help CGT developers meet future demands and identify options that will be suitable to regulators and clinicians.
Challenges remain with manufacturing reagents and consumables suitable for CGT development. Smaller volume reagents are typically not provided in closed formats. And consumables are often hand-assembled, which can lead to manufacturing inconsistency and failures. Though most culture media still come in bottles, bags with weldable lines are now becoming more common.
Despite the wider availability of ADCF reagents, off-the-shelf reagents might not always support specific cell populations of interest in a cost-efficient manner. This could motivate the development of custom media formulation. While several vendors offer custom media services, they often rely on conventional formulation techniques which typically lead, at best, to incremental improvements on current formulations.
Novel methods for media formulation involve the use of metabolomics- and proteomics-based discovery combined with design-of-experiments (DoE) approaches and the use of liquid handling robotics to allow thousands of formulations to be tested in parallel. This capability will drive both a more complete understanding of the underlying biology of CGTs and a reduction in the cost of goods.
Clinical and logistical considerations
For CGTs, the activities at a manufacturing facility form the central component of the “vein-to-vein” process. CGT developers must align manufacturing decisions with clinical and logistical constraints.
On the front end, acquiring the input cell material often depends on processes outside of the manufacturer’s direct control. These processes can include obtaining autologous cell material, such as apheresis units, from collection sites. Scheduling and sophistication can vary between sites. Similarly, handling the manufactured CGTs at clinical sites is both challenging to standardize and subject to variability of both process and analytics.
CDMOs have experience interfacing with these sites and identifying solutions to minimize risks. Typically, manufacturers aim to formulate CGTs to minimize point-of-care (PoC) manipulations. This might include selecting a cryoprotectant that is safe for direct infusion to avoid the need for wash or dilution steps. Another example is developing processes that use automated cell thawing systems.
The logistical considerations of access and shipping weigh into business decisions around using a centralized, regional, or local manufacturing model. This, in turn, will influence which CDMO(s) a therapeutic developer should work with. The proximity of manufacturing facilities to clinical sites will constrain the required shelf life of the CGT. For logistical reasons, frozen products and frozen input cell materials are much easier to manage and schedule. However, shipping cryopreserved products introduces the need for rigorous cold chain management.
Patient-specific CGTs add complexity around management of chain of custody throughout the end-to-end manufacturing process. Autologous therapies also motivate the use of digital tools to support these needs. As the CGT field matures, CDMOs are serving not only as development experts but also as supply chain, digital management, and logistical partners. Initiating plans early in the development process will set CGT developers up for success through their clinical and commercial programs.
Conclusions
It is important for CGT developers to think about their commercial manufacturing needs from the outset, starting in their preclinical development activities. CGT developers must drive the underlying biological understanding of their product, define their target product profile, and plan their clinical program. CDMOs can support these activities by working in partnership, bringing complementary knowledge and skills around process development and manufacturing. In a quickly maturing field, CDMOs are rapidly evolving as full-service partners for CGT developers by combining historical expertise from adjacent industries with new tools and capabilities specific to the unique complexities of CGTs.
By developing a strong partnership with the right CDMO at the preclinical stage, CGT developers can chart out a manufacturing development program that aligns with their clinical plans and financing expectations. This ensures that their process and analytic methods are meeting all stakeholder needs, from preclinical development all the way through to commercialization.
Download the Business of Biotech podcast series to hear from guests who turned biotherapy ideas into clinical realities.
References
1. Medvec, A. R. et al. Improved expansion and in vivo function of patient T cells by a serum-free medium. Mol. Ther. Methods Clin. Dev. 8, 65–74 (2017).