文章灵感源于 Tapas 和 TECH Talks 数字活动,其中包含从客户合作中获得的见解。专家来自 Cytiva 和 CCRM。
本文讨论了通过质量控制(QC)、优化、扩展、关闭、自动化和分析开发将风险降至最低的细胞治疗工艺开发和验证选项,以便为成功的商业生产做准备。
细胞疗法扩展策略
为了取得商业上的成功,开发的工艺必须可扩展并且适合制造环境。扩展策略取决于细胞疗法的类型。异体疗法旨在为许多患者服务。传统生物工艺处理的这种扩展策略,例如单克隆抗体治疗工艺,通常使用种子培养方法,您可以在其中跨系统扩展,逐渐增加规模。对于同种异体细胞疗法,为了最大限度地减少更珍贵的患者衍生细胞的无菌性和处理风险,需要在同一系统内进行扩展。这可以通过低细胞体积来完成,从分批进料开始,然后通过灌注提高进料和废物的产生率,以逐步扩大细胞。此外,相比之下,自体细胞疗法是针对每位患者进行的个性化治疗,因此批量较小。自体疗法使用横向扩展策略,其中添加额外的小型生物反应器以增加产量通量。
异基因工艺可在 100 mL 至 2 L 的规模上进行优化,在 Cytiva 的 Xuri 波浪式生物反应器中可实现高达 25 L 的体积。Cytiva 的 Xcellerex 系列搅拌釜式生物反应器可满足高达 2000 L 的超大批量需求。对于自体工艺,0.5 到 5 L 的体积范围通常就足够了。大型生物反应器可用于需要饲养层或刺激细胞系的其他免疫治疗平台。
CAR T 细胞制造的现代扩展策略,特别是悬浮 T 细胞培养物的扩展,非常适合转移到生物反应器中进行扩展。这允许使用具有封闭连接的一次性设备,优选无菌的可焊接管路,以及自动液体处理和灌注。与人工、劳动密集型处理相比,这些优势大大降低了风险和成本,同时最大限度地减少了操作员的操作。
CAR T 细胞工艺开发
要验证生物制品的工艺并实现生物制剂的一致生产,您必须在整个工艺过程中保持关键工艺参数 (CPP) 和关键质量属性 (CQA) 的范围。为此,必须首先仔细定义 CQA 和 CPP 并确定其优先级(1)。
除了工艺开发,尽量减少工艺差异是关键。为使细胞疗法能够在一个工艺中始终如一地测量不同患者的差异,正确的分析是必须的。
提示:在早期开发中,可通过多次测量进行重度表征来帮助您查明差异的来源并真正定义您的 CQA 和 CPP 及其范围。也可以围绕这些建立试验验收标准,以控制所需 CQA/CPP 目标输出的一致性。
为了制定最终的工艺控制和批次放行分析,您当然必须遵守法规要求,并将任何特定的物流需求纳入您的工艺。理想状态是能够展示控制,并在可行的情况下结合在线实时监控工具和技术来进行使用无损采样的分析。最终,如果您具备灵活的工艺,可以通过实时监控调整参数,您就可以简化工艺控制并确保每个患者批次能够达到目标治疗剂量和特征。
工艺开发决策
表 1 总结了在开发 CAR T 细胞工艺的商业规模制造时要考虑的关键选项。
表 1:商业规模的 CAR T 细胞生产的主要决策
| PD 关注领域 |
最佳实践 |
获益 |
||||
| ↑ 产品开发 |
↑ MFG 操作 |
↓ 工艺差异 |
↓ 生产风险 |
↓ 监管负担 |
||
|
表征 |
小规模优化 | x | x | |||
| 开发有意义的 CQA 和 CPP | x | x | x | x | ||
| 表征工艺差异的高风险源 | x | x | x | x | ||
| 将 CQA 与差异相关联以定义工艺公差 | x | x | x | x | ||
|
原料选择 |
GMP 试剂和培养基配方筛选 | x | x | x | ||
| 定义辅助试剂 | x | x | x | |||
| 过时的、定义不明确的原材料 | x | x | x | x | ||
| 使用备份识别供应商采购 | x | x | ||||
|
细胞或基因进料 |
定义输入细胞源选择和可用性 | x | x | x | ||
| 优化 T 细胞富集和分离策略 | ||||||
| 病毒载体选择和滴度优化 | x | x | x | x | ||
|
最终工艺 |
具有内置单元操作公差的 SOP 生成 | x | x | x | x | |
| 工程与集成单元操作同时运行 | x | x | ||||
| 技术转让给 CMO 以进行工艺验证 | x | x | ||||
| 工艺验证活动 | x | x | ||||
每个工艺开发方面都有不同的好处,可能会推动您关注的领域。加大对工艺开发的投资可以增加商业生产成功的机会,其中许多好处还意味着商品成本 (COG) 的降低。但最终,这必须与加快完成临床试验相平衡。我们强烈建议关闭和自动化您的制造工艺。不同的工艺有不同的需求,需要定制解决方案以适应和平衡所需的成本和时间表。Cytiva 的 Fast Trak 工艺开发服务可以帮助您在想要关注的领域实施最佳实践。
图 1 显示了如果您要完全关闭并完全自动化该工艺,间充质干细胞 (MSC) 的成本节约预测(2)。
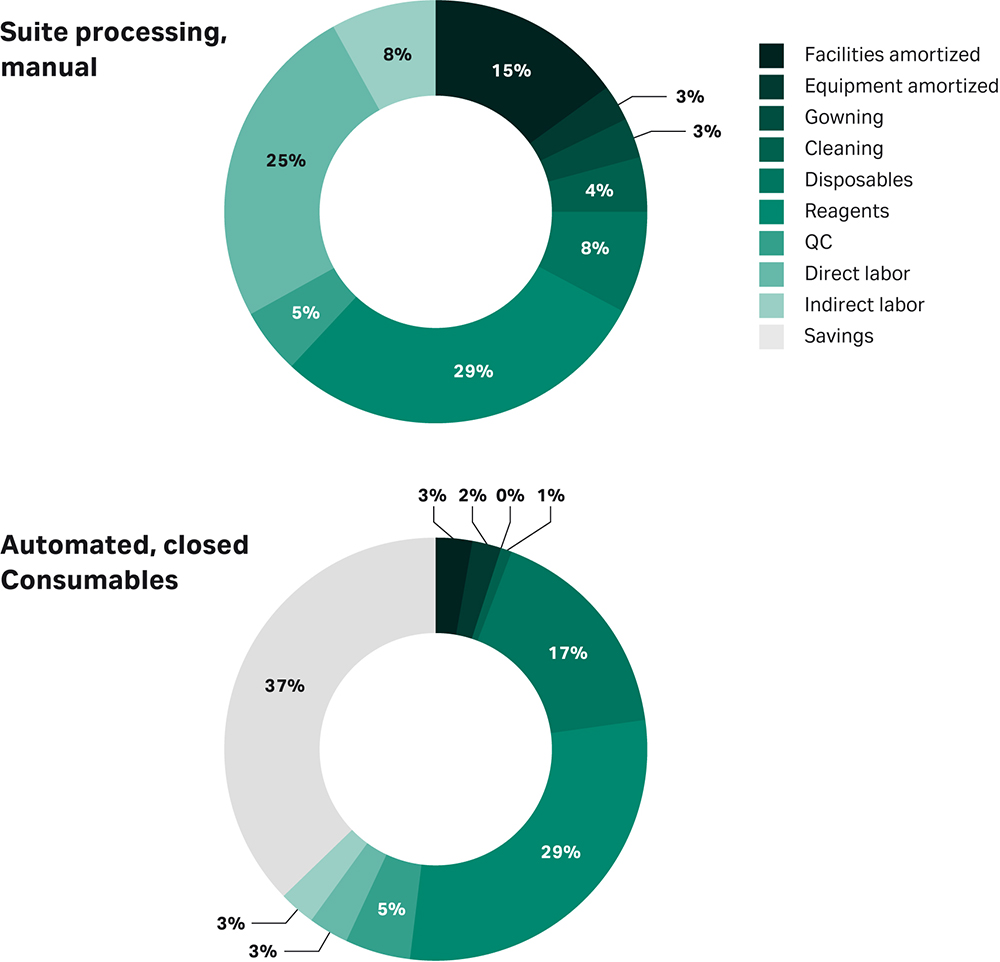
图 1:手动与封闭和自动化细胞疗法制造工艺的预测成本。此示例适用于基于 Lipsitz 等人的 MSC。细胞疗法,2017。
您越早投资于封闭和自动化处理,工艺优化就越容易,启动临床试验的技术转移也就越快。当然,您还可以简化商业运营。与容易出错的手动处理相比,自动化允许通过设备中受控和可追溯的数字输入来调整参数。此外,完全封闭的流程明显降低了污染风险,同时让您在物流方面更灵活地满足您的设施要求。
质量控制和放行测试
监管机构如何看待监控制造流程表现的重要性?他们围绕身份、强度、质量、纯度和效力使用特定的语言和术语。考虑到这一点,以及临床试验从最初关注安全性开始的事实,我们可以定义所需的要素。
从监管的角度看,其他重要方面是方法确认和方法验证。国际人用药品技术要求协调委员会 (ICH) 有一套良好、丰富的指南。补充这些的是美国药典 (USP) 关于验证、确认和方法转移的章节。如果您在美国上市,您必须要查看这些以及具体的食品药品监督管理局 (FDA) 指南。表 2 列出了关键指南,表 3 总结了第 1 阶段和第 2 阶段所需的监管类别和活动。
表 2:细胞疗法在美国上市的主要监管指南
| 机构 |
指南 |
| ICH | Q2(R1) 分析程序的验证 |
| USP | <1225>:药典方法的验证 |
| USP | <1226>:药典方法的验证 |
| USP | <1224>:分析程序的转移 |
| FDA | 药物和生物制品的分析程序和方法验证 (2015) |
| FDA | 行业生物分析方法验证指南 (2018) |
表 3:第 1 阶段和第 2 阶段所需的监管类别和活动总结
| 监管类别 |
测试材料来源 |
示例 |
所需的活动(第一/第二阶段) |
| 安全/纯度 | 药典 | 无菌性,USP <71> | 确认 |
| 内毒素,USP <85> | 确认 | ||
| 支原体,USP <63> | 确认 | ||
| 杂质检测 | 资格认证/验证 | ||
| 标识 | 工艺开发 | 流式细胞仪 | 资格认证/验证 |
| 核型 | 资格认证/仅供参考 | ||
| STR | 资格认证/仅供参考 | ||
| PCR 方法 | 资格认证/验证 | ||
| 规格 | 工艺开发 | 细胞计数/活力 | 资格认证/验证 |
| 质量 | 工艺开发 | 性能测试 | 资格认证/验证 |
| 扩散测试 | 资格认证/验证 | ||
| 动物模型 | 资格认证/验证 | ||
| 效力 | 工艺开发 | 流式细胞仪 | 资格认证/验证 |
对于安全和纯度,这些方法属于药典级别,几乎适用于所有产品。因此,只需要对无菌性、内毒素和支原体进行验证。然而,任何特定工艺的杂质都需要确认和验证。
工艺稳健性
工艺稳健性是重要的可制造性参数,可确保该工艺不仅能在完美研发空间中工作,而且还能在生产操作的正常可重复条件下工作。例如,如果必须立即收集和测试样本,这是否意味着实验室分析员必须在抽取样本时 24 小时轮班工作?目标是进行稳健性研究并证明样品在工作台、冰箱或冰柜中的稳定性,直到可以按照预定时间表进行分析的早晨。
提示:资格认证实际上是第三阶段之前方法验证的一个子集。最终,这些方法将需要全面验证,因此如果早期迹象表明可能存在影响验证的问题,则可能需要采用新策略。
总结
我们已经描述了为制造做准备的要求。虽然早期项目不需要制造工艺验证,但它确实适用于早期阶段的分析。
提示:在设计工艺时做出决策非常重要,因此在整个开发生命周期中都有指导决策制定的路径。
工艺认证的设计和评估总是尽可能地确定和减少差异,以支持商业制造的最终目标。当您完成所有这些开发工作后,您仍然可以在产品生命周期和项目推进中监控并继续改进差异并降低故障风险。
大多数细胞疗法仍然使用危险且昂贵的开放式人工过程。目标是转向封闭和自动化设备,以降低成本和风险,并实现现代化以满足监管和供应需求。
参考文献
- Lipsitz YY, Timmins NE, Zandstra PW. Quality cell therapy manufacturing by design. Nat Biotechnol. 2016l34:393-400. doi: 10.1038/nbt.3525.
- Lipsitz YY, Milligan WD, Fitzpatrick I, et al. A roadmap for cost-of-goods planning to guide economic production of cell therapy products. Cytotherapy. 2017;19:1383-1391. doi: 10.1016/j.jcyt.2017.06.009.