尽管核酸扩增检测 (NAAT) 技术取得了诸多进展,PCR 仍是所有核酸实验室的核心技术。从基础研究到应用研究,PCR 可用于识别、测序或修饰 RNA 和 DNA。
然而,即便是这一成熟方法也可能出现问题。PCR 可能产生非特异性扩增、假阳性、包含不同突变的混合产物,甚至完全无扩增产物。如何在实验中尽量避免这些挑战?PCR 的成功依赖于方法、设计、试剂和仪器的恰当选择。
尝试采用以下 PCR 设计与实施最佳实践,提升您的实验数据质量。
1. 选择最合适的 PCR 方法
PCR 有众多可选方案,包括不同的检测方法,因此很容易让您忽视更适合您应用场景的选项。
常规 PCR
常规 PCR 是一种在反应末端通过嵌入染料(如溴化乙锭或 SYBR™ Green)检测 DNA 存在与否的方法。
作为一种简单且低成本的技术,常规 PCR 可用于克隆或医学中的微生物检测,但它在现代基因组学中同样发挥作用,例如为二代测序 (NGS) 扩增起始材料。
定量 PCR (qPCR)
定量 PCR (qPCR),也称为实时 PCR (real-time PCR),通过使用荧光探针,可实现对 DNA 扩增的快速、特异性检测。
借助多重 PCR,您还可以使用不同的引物和探针识别多个 DNA 靶标。使用质量良好的样品在一次反应中同时检测多个 DNA 区域,有助于节省成本,并减少假阴性结果的发生。
对于 RNA,您可以采用一步法或两步法进行定量分析。然而,考虑到多个样品之间可能存在质量差异,一步法更难进行样品间比较,也无法重复失败的 PCR 实验。
表 1.一步法与两步法逆转录 PCR (RT-qPCR) 的优缺点汇总
| 优点 | 缺点 | |
|---|---|---|
| 一步法 RT-qPCR |
|
|
| 两步法 RT-qPCR |
|
|
这两种方法都依赖于高质量且一致性良好的样品提取,理想状态下可减少花在调整和优化逆转录反应上的时间。
直接 RT-PCR 可应对这一挑战,实现无需提取的 NAAT。
数字 PCR
数字 PCR (dPCR) 是 PCR 方法的最新进展之一。该方法将 PCR 溶液分隔为数万个纳升级的液滴,每个液滴中进行独立的 PCR 反应。
数字 PCR 能够检测更小的变异或低频事件,且比 qPCR 更能抵抗抑制剂的影响。它特别适合检测罕见突变、SNP 或基因编辑事件。但由于不依赖建立标准曲线,相较于 qPCR,它不太适合用于病原体检测或基因表达研究。
2. 制备高质量核酸
PCR 的质量与所用核酸的质量密切相关。自制试剂可以获得良好的提取质量,但商用试剂盒能减少操作差异,并标准化提取工艺,即便是微量样品也能获得高收率。
无论使用哪种方法,都可能会带入影响或抑制 PCR 的杂质。例如,苯酚会降解聚合酶,而从 Tris-EDTA (TE) 缓冲液中带入的乙二胺四乙酸 (EDTA) 可能螯合反应中的镁离子,从而降低聚合酶活性。
低质量的提取方法还可能残留核酸酶,导致样品降解。因此,记得使用分光光度法检测 A260/280 比值:纯 RNA 应约为 2.0,纯 DNA 应约为 1.8。若该比值偏低,可能表示存在盐、苯酚或蛋白质等杂质。
优化提取步骤、选择合适的洗脱缓冲液,有助于应对上述挑战。
您还可以通过直接 RT-PCR 解决核酸提取相关问题。该方法无需单独的核酸提取步骤,不仅可缩短检测时间、提升通量,还能实现从未纯化样品中快速、准确、灵敏地检测病毒 RNA,例如从鼻咽拭子中检测 SARS-CoV-2 病毒 RNA。
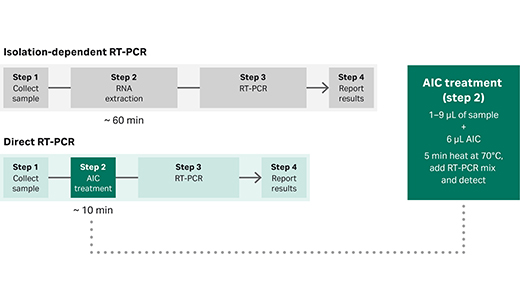
图 1.依赖核酸提取的 RT-PCR 与直接 RT-PCR 的流程比较。
我们的直接 RT-qPCR 主混液试剂盒包含抗抑制剂复合物 (AIC),可在裂解样品的同时保护 RNA 完整性(图 1)。
了解直接 RT-qPCR 主试剂盒,实现免提取 RNA 分析
3. 花时间设计合适的引物和探针
设计理想的引物并不简单。遵循一些基本规则有助于避免引物自退火、形成引物二聚体,或引物对间的熔解温度 (Tm) 差异过大等问题。
多种在线引物设计工具可帮助您识别序列中可能存在的这些问题。
那么,引物设计的规则有哪些?
理想引物应具备以下特征:
- 为短链 (15-30 kb) 的寡核苷酸序列。
- 对靶区具有高度特异性。
- 重复核苷酸序列极少。
- GC 含量较低(< />
适应高 GC 含量
鸟嘌呤-胞嘧啶 (GC) 含量是重要的考量因素,因为 GC 含量高会导致键合稳定性增加,会提高引物的熔点,从而容易引发二级退火。
如果模板本身富含 GC,可选择专为此类模板优化的反应主混液。这类主混液通常含有二甲基亚砜 (DMSO) 等添加剂,并配备优化的聚合酶。
利用剪接位点
在研究 RNA 时,需先合成 cDNA。但如何避免扩增样品中污染的 DNA?
虽然可以使用 DNase I 等核酸酶处理样品,但这种方法可能无法完全去除 DNA 污染。因此,您可以考虑设计跨越外显子-外显子连接位点的引物。这种设计能确保只扩增 cDNA,同时也便于分析特定的剪接变体。
同样地,在研究 DNA 时,也可设计跨越外显子-内含子连接位点的引物,结合使用 RNase,以避免扩增污染的 RNA。
4. 寻找最适用的探针
嵌入染料
使用嵌入染料(如经典的溴化乙锭)染色凝胶,是检测常规 PCR 的常用方法。溴化乙锭成本低、灵敏度高,且操作简便。
不过,使用嵌入染料时需要注意一个问题:当目标模板丰度较低时,可能会形成非特异性产物,从而增加背景信号。以 SYBR Green 为例,当浓度超过一定阈值时,该染料还可能抑制 PCR 反应。
一种可替代的选择是 EvaGreen™ 染料,它经过设计,具有较低的背景信号和更高的信号强度,同时对 PCR 的抑制作用也更小。这些特性使其非常适用于高分辨率基因分型等高灵敏度应用。
荧光探针
在设计 qPCR 实验时,您可以选择多种带荧光标记的寡核苷酸探针,包括水解探针、分子信标探针以及双重杂交探针。在所有类型中,探针应靠近引物之一,且 Tm应比引物略高(高出 6-8°C),同时具有较低的 GC 含量。
那么,对于 qPCR 而言,嵌入染料和荧光探针哪种更好?可参考表 2 中的优缺点比较。
尽管荧光探针的设计相对复杂,但各种在线工具能大大简化此过程。您可以根据 qPCR 循环仪制造商为特定仪器所校准的染料选择,设计双工实验。例如,发射波长为 517 nm 的 6-FAM 标记探针可与 Cy™ 3 (564 nm) 或 Cy™ 5 (670 nm) 标记探针复用。但若将 6-JOE (548 nm) 标记的探针与 Cy™ 3 标记的另一探针复用,可能会导致信号色度亮度干扰,从而影响数据分析的准确性。
表 2.qPCR 中嵌入染料与荧光探针的优缺点比较
| 优点 | 缺点 | |
|---|---|---|
| 嵌入染料 |
|
|
| 荧光探针 |
|
|
5. 请勿忽视反应中的其他试剂!
主混液试剂盒已对 DNA 扩增的基本组分进行了预优化,但当 PCR 反应失败时,仍有必要逐一评估各试剂对特定反应体系的影响。
典型的主混液包含:
- DNA 聚合酶
- dNTPs
- MgCl2
- 缓冲液(含任何添加剂)
DNA 聚合酶
Taq DNA 聚合酶是最常用的一种聚合酶,广泛应用于各种常规 PCR 实验。尽管缺乏校对能力,但 Taq 聚合酶仍是 qPCR 实验的良好起点。
但如果您要扩增 GC 含量较高的区域,可尝试专为此类难扩区域设计的聚合酶,例如 Pfu DNA 聚合酶。
同样地,如果遇到非特异性扩增的问题,可以选择热启动型聚合酶。这类聚合酶在低于其最佳反应温度时处于非活性状态,有助于在设置阶段减少非特异性扩增。
dNTPs
在常规反应中,核苷酸通常等摩尔加入。但在随机诱变等应用中,可能会使用不平衡的 dNTP 浓度以促进错误插入。
在常规使用中,dNTP 的推荐浓度为 0.2 mM。若需提高MgCl2浓度,则应相应提高 dNTP 浓度。但需确保主混液中的 dNTP 浓度不低于 0.01 mM,以免影响 PCR 扩增效率。
MgCl2
镁是 DNA 聚合酶和引物杂交的关键辅助因子。因此,可通过调整其浓度来优化 PCR 效率。
推荐初始浓度范围为 1-4 mM,随后按梯度微调。镁浓度过低可能导致 PCR 扩增产物极少或无产物;而浓度过高则可能导致反应中出现非特异性产物。
缓冲液和添加剂
缓冲液为 DNA 聚合酶提供适宜的化学反应环境,并能减少 DNA 损伤。
例如,二甲基亚砜 (DMSO) 是常见的添加剂之一,可用于降低 GC 含量高区域引发的二级结构的形成。
6. 提高 GC 含量区域的扩增效率
为便于扩增 GC 含量高的区域,可尝试以下几种方式:
- 提高熔解温度(但需注意,聚合酶在超过 95°C 时可能会加速变性)。
- 通过MgCl2滴定进行 PCR,优化目标序列的最佳浓度。
- 添加有助于减少二级结构形成的添加剂,例如:
- 3–10% DMSO
- 1.25% 甲酰胺
- 5% DMSO 和 1.25% 甲酰胺
- 甜菜碱(终浓度 2.2 M)
- 乙二醇(终浓度 1.075 M)
- 1,2-丙二醇(终浓度 0.816 M)
此外,更简便的方法是选择针对难扩区域优化的稳健商业化主混液试剂,例如 Cytiva 的 RT-qPCR 试剂盒。此类主混液含有经过优化的试剂组合,可显著提高扩增效率。
以我们的产品为例,Cytiva RT-qPCR 主混液采用冻干形式提供,便于储存和使用。
7. 设置合适的对照组
确保实验中设置了所有必要的对照,包括:
- 阴性对照:例如,无核酸酶的水,替代模板 DNA。
- 阳性对照:已知含有目标序列的样品。
- 无 RT 对照:用于评估样品中是否存在污染性核酸。
- 管家基因:作为内源性参考,用于校正样品间的差异。
如果您正在排查反应失败的原因,在样品中加入外源阳性对照是一种有效方法,可用于判断是否存在影响 PCR 的抑制因子。
8. 检查可用的设备和耗材
热循环仪在 PCR 循环的每一步中负责对样品温度的精准和可重复升降温操作。这些仪器可能采用加热模块或热风系统,实现对反应管的快速加热与冷却。
不同型号在升温速率和温度均一性方面的性能各不相同,因此务必确认其是否符合您的实验需求。例如,升温速率可能会影响非特异性产物的生成。
部分 PCR 仪器配备梯度功能,可用于优化每个目标反应的温度设置。您可以评估整个模块的温度均一性,以判断其是否会影响实验结果,因为模块边缘较低的温度可能导致 PCR 结果出现差异。
最佳实践助力可靠 PCR 结果
尽管 PCR 技术已有数十年历史,科学家仍在不断拓展其应用边界。请参考我们的最佳实践建议,设计并优化您的 PCR 以实现最佳结果。如果您遇到意料之外的实验结果,也可回顾此清单,确保已为 PCR 成功做出最合适的选择。
在 Cytiva,我们提供广泛的 DNA 样品制备和 PCR 解决方案。如需了解最新动态、技巧和见解,欢迎访问我们的基因组学博客。如您在实验流程中任何环节需要支持,请联系我们的科学支持团队。