什么是生物制药的工艺开发?
上游或下游生物工艺开发的定义、活动以及需要考虑的事项。
什么是生物制药的工艺开发?
生物制药工艺开发包括生产生物分子的一系列步骤 – 单克隆抗体 (mAb)、重组蛋白、病毒载体或其他生物来源的产品。
生物工艺开发通常分为上游工艺开发和下游工艺开发。这些必须与恰当的分析方法相结合,只有这样,您在工艺开发和优化过程中才能对定义的产品关键质量属性 (CQA) 进行准确测定。
在药物开发的早期阶段,您将开发出一个足以满足该阶段需求的“足够好”的工艺。然而,牢记最终目标非常重要。最终,您将需要一个能转换为商业化生产的工艺,即在整个临床试验和上市过程中易于放大的工艺。当您进入药物开发的临床 III 期阶段时,只是“足够好”是不够的。与此同时,您还需要将注意力转移到确保建立稳健的上游或下游工艺上来,以保证获得高收率和高生产率。此外,工艺还必须注重成本效益和可重现性。
牢记目标,确保未来取得成功
早期关键决策的制定将对目标的实现产生重要影响。例如,与贴壁细胞相比,悬浮细胞培养更容易放大。在工艺放大过程中,通过超速离心来分离腺相关病毒 (AAV) 的空衣壳和完整衣壳的效果并不理想。应从一开始就考虑工艺的可放大性,以避免后期返工。
如果您在临床试验期间对生产工艺进行变更,这可能会影响到 CMC(化学、制造和控制)方面监管材料的提交,并可能会对后续的项目时间表产生负面影响。因此,如果您希望获得长期成功,尽早做好工艺设计非常重要。
当您正在开发一种生物分子,并且很有可能会获得监管机构的快速批准,则尽早做好工艺设计尤为重要。如果尽早制定工艺开发、质量保证和质量控制策略方面的关键决策,可以为cGMP生产做好准备。同时集中精力确保生产工艺的稳健性。
工艺开发必须平衡产品质量和工艺(图 1)。市场对速度和经济性的追求正在驱动尽早决策,以及加速产品上市速度以获得更理想的价格。这些需求都转化为更大的压力,即快速开发出注重成本效益且具有生产规模的工艺。这些综合因素可以帮助提高生产率,从而对工艺的经济性产生巨大的积极影响。
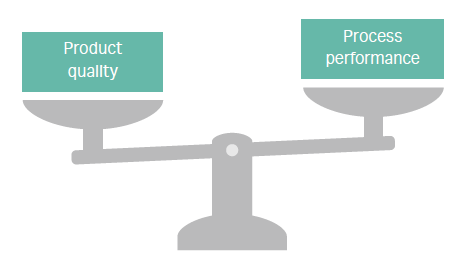
图 1. 生物制药工艺开发是一个注重平衡的过程。
希望了解 CMC、cGMP 和其他关键术语?请查看我们的生物工艺开发定义。
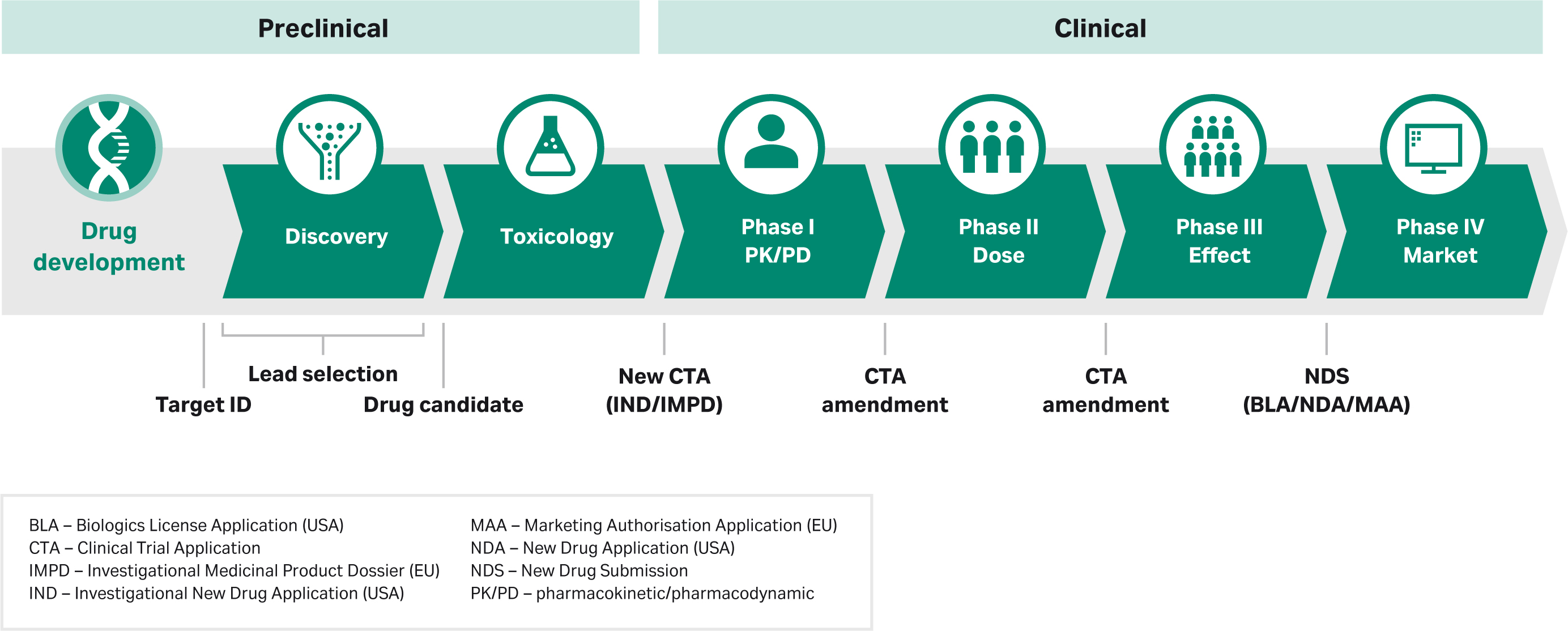
药物开发过程各阶段的工艺开发程度不同。虽然世界各地各阶段的基本进度相似,但各地区的具体内容不同,具体取决于相关监管机构的指南。
毒理学和早期临床阶段的工艺开发:关注重点工作
该早期阶段的工艺开发工作对于确保产品的长期稳健性非常关键。此阶段的重点工作是确定几个重要参数。您的工作必须为毒理学 (tox) 和 I/II 期研究的产品生产提供支持,并为监管申报提供支持。您的目标是开发具有中间收率的工艺,同时满足所有产品质量期望。该第一版工艺采用初始产品质量规格进行开发。开发的工艺应具有放大为大规模生产的潜力,而且不必进行大的调整。
为使工艺开发活动以确定关键工艺参数作为重点,您需要结合以往的知识与经验分步骤地确定这些参数,并帮助确定需要优先开展的实验。您可以使用鱼骨图帮助确定工艺参数。为了确定需要优先开展的活动,您可以使用优先级矩阵或失效模式和影响分析 (FMEA)。
在下游工艺结束时,您将获得原料药 (DS)。然后将原料药配制成制剂 (DP)。确保尽早开始预配制活动。在此阶段应保持简单的制剂 (DP) 配方;或许可以将其配制成冷冻溶液。
III 期和商业化生产的工艺开发:确保工艺的稳健性
在此阶段,您的工艺必须适合进行商业化生产。由于工艺开发的工作量较大,一般采用实验设计 (DoE) 研究。此时您的主要工作是按照质量源于设计 (QbD) 的原则定义设计空间。您需要通过工艺表征优化并开展大量的工作确定关键工艺参数 (CPP)。总之,您需要关注稳健性、收率、可重现性和可放大性,最终获得经济高效的工艺。
在临床试验的后期阶段,您需要开发上市的制剂 (DP) 配方。例如,您可能需要考虑采用冻干技术延长有效期。在监管申报阶段,您需要展示 I 期与 III 期产品的可比性。这样做的原因是向监管机构证明产品未随着时间的推移发生变化,并且临床试验结果具有代表性,对于商业化产品具有支持作用。
关于早期和后期的详细活动分解,请参阅以下部分:
上游工艺开发
下游工艺开发
希望了解 IND、CMC 和其他关键术语?请查看我们的生物工艺开发定义。
上游工艺开发是指在收获纯化前从细胞系开发一直到生物反应器工艺开发整个流程。细胞系开发 (CLD) 是生物制造工艺中的一个关键步骤(例外情况:在自体细胞疗法中,起始物料的来源是患者自身细胞)。CLD 的进步提高了滴度,并使工艺开发者能够更快地将分子投入到临床研究。不同疗法可能使用不同的宿主细胞系,但在任何一种情况下都需要单个克隆。
生产系统取决于生物分子类型。例如,如果待生产抗体片段不需要特定的翻译后修饰(例如糖基化模式),可以选择微生物系统。另一方面,如果必须使用哺乳动物系统,可以选择许多来源于中国仓鼠卵巢 (CHO)、人类胚胎肾 (HEK) 或其他哺乳动物细胞的特定细胞系。昆虫细胞适合生产某些生物分子。细胞培养基平台开发是上游工艺开发的另一个重要步骤。对于任何一种工艺,从监管角度来看,理想情况是使用无动物源性成分(即 ADCF)的培养基和进料,因为这样可以降低动物源性外源病毒的潜在风险。
上游工艺开发包括:
在您开始 I/II 期开发前
早期上游工艺开发从生成特定细胞系和选择单个克隆开始。此间您将通过实验室规模的试验对生长情况、稳定性和生产系统生产率进行评估。
重要的是在开展毒理学 (tox) 研究和 I/II 期生产活动之前,留出生产符合药品生产质量管理规范 (GMP) 要求的细胞库的时间。应确保您对亲本细胞系和载体拥有自由实施权或商业许可。
在您开始 III 期开发过程前
后期上游工艺开发的重点是定义和优化关键参数,以提高生产率、工艺稳健性和产品质量。为实现生物制品的生产一致性,您必须在整个开发过程中将关键工艺参数 (CPP) 和关键质量属性 (CQA) 维持在区间范围。为此,您必须了解您的 CQA,确定 CPP 及其范围,并了解与工艺相关的风险。您还必须为商业化生产制定相关的风险缓解策略。为满足产品质量规格的要求,设计空间需定义 CPP 的可接受范围,这也是质量源于设计 (QbD) 工具的一个关键方面。
表 1. 按临床阶段划分的上游工艺开发活动汇总
| 上游工艺开发阶段 | 活动 |
| 早期临床阶段 | |
| 临床后期阶段 |
|
文献
Adapted table used with kind permission: Lindskog E., “Chapter 31 The upstream process: principal modes of operation” in G Jagschies et al., “Biopharmaceutical Processing: Development, Design, and Implementation of Manufacturing Processes”. Elsevier 2018.
希望了解 CPP、CQA 和其他关键术语?请查看我们的生物工艺开发定义。
下游工艺是指在上游工艺完成后目标分子的回收和纯化。下游工艺包括收获、纯化和病毒清除步骤。通常将多个不同步骤安排和/或组合在一起以纯化目标产品。在下游工艺开发期间对工具和工艺条件(即关键工艺参数或 CPP)进行定义。目标是在产量和质量(即关键质量属性 [CQA])之间找到适当的平衡。
首先,您可能需要以实验室规模进行工艺开发工作。为了加快工艺开发并提高效率,您可以通过高通量工艺开发 (HTPD) 对工艺条件进行初步筛选。或者,您可以使用 HTPD 对定义的空间进行更彻底的研究,为对工艺的详细理解和/或稳健性研究打下基础。针对层析工艺进行探索和筛选后,您将使用较大的层析柱进行验证和优化。以下表格按下游工艺开发阶段对实验活动进行了汇总。
表 2. 按临床阶段划分的下游工艺开发活动汇总
| 下游工艺开发阶段 | 活动 |
| 临床早期阶段 |
|
| 后期临床阶段 |
|
希望了解 CPP、HTPD 和其他关键术语?请查看我们的生物工艺开发定义。
在工艺开发的几个关键阶段和制造过程中的关键点对质量属性进行测定和控制。关键质量属性 (CQA) 无疑是监管合规性的关键。在 ICH Q8 中,对 CQA 的定义定义决定产品质量的性质或特征(即对患者至关重要的属性。)监管机构还需要获得工艺的 CQA 数据,以确保工艺步骤按照预期完成。您需要从能够表征分子的生物学功能的 CQA 开始着手。例如包括受体结合亲和力和正确的糖基化。总的来说,生物制品 ― 病毒、mAb、细胞(用于细胞疗法)、抗体-药物偶联物 (ADC)、病毒等等 ― 都是复杂的大分子,需要采用稳健且可靠的方法对其进行深入分析和表征。
以下列出了基于蛋白的生物分子典型分析。其他类型的生物疗法通常使用其中一些分析方法,不过也有自己的一些分析方法。
- 纯度
- 相似度
- 生物活性
- 结合活性
- 宿主细胞蛋白 (HCP) 和其他杂质
- 浓度
- 大小和聚集体
- 可比性
希望了解 CQA 和其他关键术语?请查看我们的生物工艺开发定义。
- 什么是生物制品许可申请 (BLA)?生物制品许可申请 (BLA) 是指向美国食品药品管理局 (FDA) 提出正式申请,申请批准新型生物药物(生物制剂)在美国销售和上市。BLA 类似于新药申请 (NDA),但由于生物疗法制造的性质,BLA 更为复杂。
- 什么是制药 CFR?CFR 是指《美国联邦法规》,其中编纂了在《美国联邦公报》中发布的规则(法律)。其中负责管辖食品和药品的部分为第 21 篇,被称为 21 CFR。
- 什么是 cGMP?在美国,现行药品生产质量管理规范 (cGMP) 是一系列关于药品或生物制品的最低方法和控制要求,目的在于确保药物或生物制品生产的安全性和有效性。“c” 用于强调所使用的系统和控制必须符合监管机构最新的期望。
- 什么是关键物料属性 (CMA)?关键物料属性 (CMA) 是指投入(原)材料的性质或特征应保持在适当的范围内,以确保产品质量符合要求。CMA 可以是物理、化学、生物学或微生物学方面的属性。例如:用于制作细胞培养基的铁载体纯度/杂质水平。
- 什么是药物开发的 CMC?化学、制造和控制 (CMC) 是美国监管申请中的一部分,其中详细介绍药物的制造工艺、如何确保产品质量以及测试方法。这些详细信息可让监管机构确信产品每次都能按照相同的高质量标准生产。
- 什么是关键工艺参数 (CPP)?关键工艺参数 (CPP) 是与制造相关的设备或工艺的特征,其差异对药物的关键质量属性 (CQA) 有影响。应对 CPP 进行监测或控制,以确保按照工艺生产的产品质量符合预期。 例如:影响生物反应器中氧传递给细胞的因素。
- 什么是关键质量属性 (CQA)?关键质量属性 (CQA) 是指对治疗产品质量具有重要影响的产品属性或特性。CQA 可以是物理、化学、生物或微生物特性或特征,但必须加以定义、测量和控制,以确保所需的产品质量。例如:嵌合抗原受体 T (CAR T) 细胞疗法的纯度 (CD34+)。
- 什么是 CTA?临床试验申请 (CTA) 是一个通用术语,指为获准在临床环境中使用研究药物而首次提交申请。CTA 的提交文件在美国为研究性新药 (IND) 申请,在欧盟为研究用药文档 (IMPD)。
- 什么是实验设计 (DoE)?实验设计 (DoE) 是一种系统化的方法,用于改变工艺的输入并分析结果,以寻找因果关系。DoE 技术可通过同时改变几个参数,最大限度地减少实验次数。
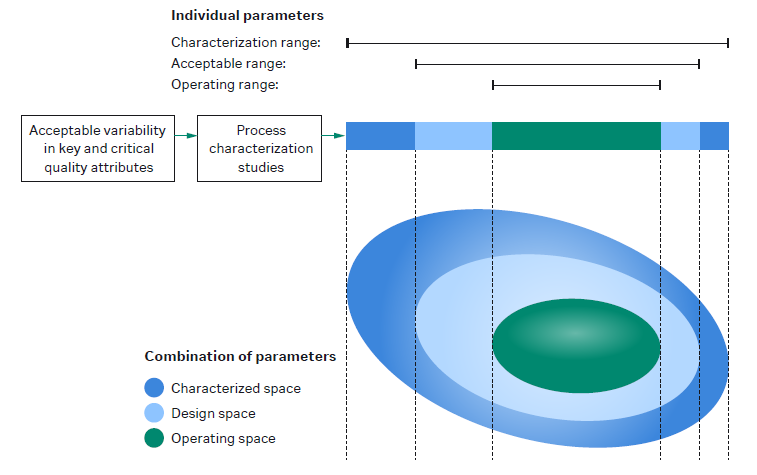
有关更多信息:观看短视频,了解蛋白纯化的 DoE。 - 什么是 QbD 的设计空间?质量源于设计 (QbD) 的设计空间是证明可保证产品质量的输入材料和工艺参数的综合相互作用。设计空间是表征范围的子集,即工艺开发过程中获得的所有知识。设计空间内定义了一个较小的控制或操作空间。
该图显示了表征空间、设计空间和操作空间之间的关系。 - 原料药和制剂之间的区别是什么?原料药 (DS) 是下游工艺结束时获得的散装物质。在灌装/成品步骤中,将 DS 配制成最终缓冲液并转移至最终容器(如小瓶)中。由此得到制剂 (DP)。
- 什么是 EMA?欧洲药品管理局 (EMA) 负责欧盟药品的科学评估、监督和安全性监测。
- 什么是 FDA?联邦药品管理局 (FDA) 是一家美国机构,负责确保人用药品、疫苗和其他生物制品以及医疗器械的安全性和有效性。
- 什么是首次人体 (FIH) 研究?首次人体 (FIH) 研究与 I 期临床研究相同。与体外或动物临床前研究相比,首次人体研究是在人体中进行的。
- 什么是药品安全性试验规范 (GLP)?药品安全性试验规范 (GLP) 描述非临床安全性研究(即动物临床前安全性研究)质量体系的监管要求。一般而言,计划、执行、监测、记录、存档和报告的方法也适用于工艺开发,通常称为 GLP。
- 什么是高通量工艺开发 (HTPD)?高通量工艺开发 (HTPD) 是一种可加快工艺开发的方法。HTPD 通常受益于自动化和多个实验条件的并行运行。可以同时评估各种实验条件,而不是等一个实验的结果出来后再开始另一个实验。HTPD 可用于表征设计空间,有助于确定需要监测和控制的工艺参数。HTPD 生成的大量实验数据创建了大量数据集。重要的是要用高通量分析法来分析这些数据集。
- 什么是 ICH?国际人用药品注册技术协调会 (ICH) 协调世界各地越来越多的监管机构采用的监管指导原则。
- 什么是 IMPD?如果计划在一个或多个欧盟成员国进行临床试验,则需要提供研究用药文档 (IMPD)。
- 什么是研究性新药申请 (IND)?研究性新药申请 (IND) 是指向美国食品药品监督管理局 (FDA) 提出申请,批准将研究性新药或生物制品用于人体。在美国,进行 I 期临床试验前需提交 IND,并根据后续临床阶段进行更新。所有地区均需提交某种类型的申请才能进行人体临床研究。不同地区的申请名称可能不同(例如,欧洲的 IMPD、加拿大的 CTA、中国的 IND 等)。
- 什么是 MMA?上市许可申请 (MAA) 是指向欧洲药品管理局申请将新药引入欧盟市场的许可。
- BLA 和 NDA 有什么区别?两者都是向美国 FDA 提出正式请求,要求批准在美国境内销售和上市新药。BLA 或生物制品许可申请适用于生物制品药物。NDA 或新药申请适用于所有其他药品(即小分子药物)。
- 什么是 NDS?新药申请 (NDS) 是通用术语,意为申请允许将新药引入市场。
- 什么是 NMPA?中国国家药品监督管理局 (NMPA) 前身为国家食品药品监督管理总局 (CFDA),是中国的药品、医疗器械和化妆品监管机构。
- 什么是药代动力学/药效学 (PK/PD) 研究?PK/PD 研究分析体内药物浓度随时间变化的规律、药物如何影响身体以及身体如何清除药物。
- 什么是生物制造平台?在这种情况下,生物制造平台是一组生产相关分子的特定设备或特定工艺。生物制造平台不是从头开始研究每种新分子,而是在以迭代方式获得的知识和经验的基础上建立的。理想情况下,平台只需要稍作改变就能适应相关分子的研究,从而节省时间和成本。
示例:随着批量变化,可放大或缩小的一系列生物反应器。 - 什么是过程分析技术 (PAT)?过程分析技术 (PAT) 是一种测量、分析和控制影响产品关键质量属性 (CQA) 的关键工艺参数 (CPP) 的方法。PAT 注重及时测量,最好是在工艺过程中进行,是使用质量源于设计 (QbD) 方法进行生物制药生产的关键组成部分。示例:使用化学光学传感器进行生物工艺过程监测,以确保符合 CQA。
- 什么是工艺表征?工艺表征研究可深入了解各工艺步骤的目的以及工艺输入对工艺输出的影响。工艺表征研究有助于调查工艺偏差。
- 什么是工艺稳健性?工艺稳健性是指工艺对规定的来源变化的耐受能力(例如投入物料的变化或工艺设备的变化),而不影响产品质量。
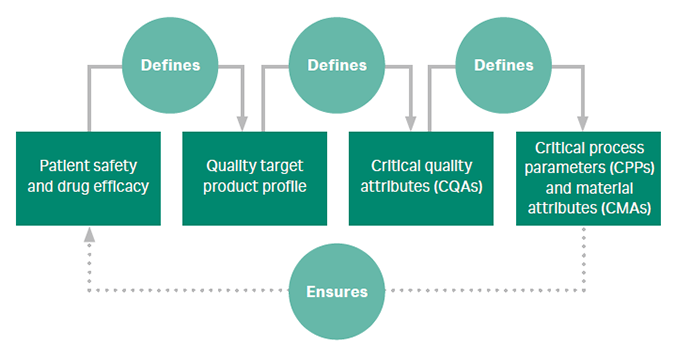
- 什么是质量源于设计 (QbD)?质量源于设计 (QbD) 是一套指导产品和工艺设计的原则,其理念是可以从一开始就“嵌入”质量。QbD 是重要的工艺开发工具,用于定义确保质量、收率和耐受变化的稳健工艺的参数。
该图汇总了 QbD 的系统方法。 - 筛选实验设计 (DoE) 可确定所使用的许多因素或条件中的哪一项对反应有显著影响。例如:通过筛选以预测阳离子交换器的步骤洗脱条件。
参考资料
U.S. Department of Health and Human Services, Food and Drug Administration. Guidance for Industry PAT — A Framework for Innovative Pharmaceutical Development, Manufacturing, and Quality Assurance. Sept. 2004. Accessed July 18, 2021.
International Conference on Harmonization (ICH) and FDA Guidance for Industry, Q8 (R2) Pharmaceutical Development, Nov. 2009. Accessed February 22, 2021.
European Medicines Agency. Quality by design. https://www.ema.europa.eu/en/human-regulatory/research-development/quality-design. Accessed February 22, 2021.
工艺开发过程中发生的情况对满足监管要求有很大影响。在本节中,我们将重点介绍美国的流程。各研究阶段的总体进展与世界其他地方相似,但具体情况因地区而异,取决于相关监管机构的指导方针,例如欧洲的欧洲药品管理局 (EMA) 或中国的中国国家药品监督管理局 (NMPA)。下图显示了美国 FDA 的监管申报在药物开发过程中的作用,并添加了一些与其他地区相关的术语:
在进行临床试验之前,体外和动物研究的药理学和毒理学数据至关重要。提交研究性新药 (IND) 申请时需要这些数据。IND 申请是一个关键里程碑,因为监管机构会对纳入的数据进行审查,以确定 1 期研究、首次人体 (FIH) 研究、试验或 1 期新治疗适应症的使用是否有充分的依据。小型 1 期研究可证实候选药物的安全性,以供进一步研究。
虽然 IND 申请需要包括 I 期方案批准所需的基本产品表征数据,但 IND 中包含的信息是随着时间的推移不断积累的。本文件将带您了解整个监管审批流程,并通过扩展纳入了各个阶段和并行进行的所有研究的数据。IND 申请包括一个涉及化学、生产和控制 (CMC) 的部分,随着时间的推移,获得的数据和工艺知识越来越多,该部分也将在整个临床试验中不断扩展。
IND 中包含的信息开发的稳定性至关重要,因为监管机构通过分析这些数据来确定药物是否有资格通过审批程序。例如,在针对受累患者的探索性 2 期研究中对几种方案进行试验之前,您可能无法最终确定配方、给药途径、适应症和给药细节。一旦这些关键指标被确定,支持最终配方、剂量、目标人群和适应症的数据才会被添加到 IND 中。在每轮监管报告中,监管机构会审查新信息,如果认为合适,就会允许候选药物进行更大规模的研究,如确证性 3 期试验。
对于一些候选药物,监管机构可以准予加快审批通道。在这种情况下,可将不同临床阶段合并进行,以加快推进速度。但监管机构仍然必须确保药物的安全性和有效性达到应有的水平。
对于中国的临床试验,NMPA 建议在 1 期、2 期和 3 期研究的设计上保留一定的灵活性,以便日后可以将 1/2 期合并为早期临床试验阶段,将 3 期作为确证性临床试验阶段。随后是 4 期,即上市后阶段。NMPA 还建议在适时遵循 ICH 指导原则。NMPA 已经发布了多份细胞和基因治疗的指导文件 – 参见参考资料部分。
与药物开发的其他方面一样,提前进行监管规划可以避免日后的麻烦。作为一项关键决策,您了解团队何时缺乏监管经验和专业知识,以及何时咨询顾问或聘请专家来填补知识空白。
为避免意外,请勿等到提交申请后再与监管机构进行接洽。可以安排几次会议来获得监管机构的反馈,例如与 FDA 的 IND 前会议。应根据 FDA 规定的程序提前征求他们的意见。
中国细胞和基因治疗产品的参考资料
National Medical Products Administration. Technical guidelines for research and evaluation of cell therapy products, 2017.
National Medical Products Administration. GMP appendix – cell therapy products, 2019.
National Medical Products Administration. Guiding principles for pharmaceutical research and evaluation of gene therapy products, 2020.
National Medical Products Administration. Technical guidelines for clinical trials of immune cell therapy products, 2020.
National Medical Products Administration. Technical guidelines for clinical trials of human stem cells and their derived cell therapeutic products, 2020.
National Medical Products Administration. Guiding principles for clinical trial design of oncolytic viruses, 2020.
希望了解 IND、CMC 和其他关键术语?请查看我们的生物工艺开发定义。
测试您的知识储备: